Xyy-синдром
Содержание:
- Диагностика
- Прогноз
- Этиология
- Распространённость заболевания
- Причины возникновения
- Клиника
- Как лечить синдром Клайнфельтера?
- Борьба с бесплодием
- Двойные аберрации
- Синдром Клайнфельтера – причины возникновения
- Диагноз
- Тактика общения с пациентами и их родителями
- Сравнительная клиническая характеристика различных видов анеуплоидий
- Тактика общения с пациентами и их родителями
- Последствия лечения синдрома Клайнфельтера
- Что такое синдром Клайнфельтера?
- Лечение
- Близкие расстройства
Диагностика
Обнаружить наличие синдрома Клайнфельтера можно на стадии эмбрионального развития при помощи скрининга биохимических показателей крови материи инвазивного пренатального исследования:
Отмечается, что чем больше лишних женских хромосом в кариотипе, тем более выражена заторможенность интеллектуального развития
- амниоцетез (срок: между 16 и 18 неделями)– забор пункцией жидкости из околоплодного пузыря;
- биопсия хориона (срок: с 9,5 до 12 недель)– анализ ворсинок зародышевой оболочки;
- кордоцентез– исследование пуповинной крови.
После забора материала из него экстрагируется ДНК будущего ребенка для изучения в лабораторных условиях на наличие синдрома. Точность вышеперечисленных диагностических процедур составляет 99%. Однако они небезопасны:
- возможен выкидыш;
- подтекание околоплодных вод;
- инфицирование;
- кровотечение.
Процент осложнений составляет 1-2%, но назначается подобная диагностика только при наличии показаний: возраст матери более 35, скрининг выявил риск, в семье есть ребенок с синдромом Клайнфельтера, будущие родители состоят в родстве.
Если синдром Клайнфельтера обнаружен, то это не является причиной для прерывания беременности. Ребенок вполне может родиться без склонности к явно выраженным признакам заболевания, а при грамотной гормональной коррекции в пубертатном возрасте не будет отличаться от здоровых. Решение остается за родителями. При повторной беременности вероятность дефектной комбинации хромосом 1%.
После рождения ребенка диагностику осуществляют эндокринологи, генетики и андрологи. При наличии синдрома Клайнфельтера в слизистых оболочках ротовой полости можно обнаружить тельца Барра, что является достаточно достоверным показателем присутствия патологии.
Выявить синдром Клайнфельтера могут следующие методы обследования:
- УЗИ мошонки покажет, есть ли аномальные изменения плотности и размеров яичек;
- анализ крови на гормоны: при наличии патологии уровень тестостерона понижен, а количество лютеинизирующего и фолликулостимулирующего гормонов повышен;
- исследование спермограммы на наличие жизнеспособных сперматозоидов;
- при помощи биопсии яичек выявляется гиалиноз канальцев, гиперплазию клеток Лейдига (источников тестостерона) и уменьшение объема клеток Сертоли, питающих сперматозоиды и вырабатывающих поддерживающие сперматогенез гормоны.

Выявить синдром Клайнфельтера может анализ крови на гормоны
Но самым достоверным методом является анализ кариотипа (кариотипирование). В ряде случаев при невыраженных клинических признаках синдром Клайнфельтера остается нераспознанным пожизненно: мужчина обращается к различным врачам с заболеваниями (проблемами бесплодия, ожирения), причины которых не удается выявить, лечение при этом часто неэффективно.
Прогноз
Младенцы с формой Клайнфельтера 47, XXY мало отличаются от здоровых детей. Результаты одного исследования, не являющиеся мозаичных XXY детей в возрасте до 2-х лет показали, что большинство XXY детей имели нормальные наружные половые органы и черты лица с высотой и весом в пределах нормы.
Мальчики с кариотипом 47, XXY могут испытывать в подростковом возрасте трудности с учебой, различные фрустрация и, в некоторых случаях, серьезные эмоциональные или поведенческие трудности. Однако большинство из них, вступая во взрослую жизнь, стремятся к полной независимости от своих семей. Некоторые закончили высшее образование и имеют нормальный уровень жизнедеятельности. Синдром Клайнфельтера не влияет на продолжительность жизни.
Этиология
Синдром Клайнфельтера обусловлен мутацией генов, приводящей к удвоению женской половой хромосомы в кариотипе мужчины. Нерасхождение половых хромосом в процессе мейоза либо митоза может быть обусловлено различными факторами. Самыми распространенными среди них являются:
- Вирусы,
- Неполноценность иммунной системы матери или отца,
- Плохое экологическое состояние окружающей среды,
- Дети от родственных браков,
- Ранний или поздний возраст матери,
- Наследственные патологии в предыдущих поколениях.
Мальчики с синдром Клайнфельтера вместо нормального мужского генотипа ХУ приобретают одну У хромосому и несколько Х хромосом. Такое изменение генетического набора приводит к появлению особых внешних данных, незначительному снижению интеллекта и развитию целого ряда сопутствующих заболеваний.
Повышение концентрации фолликулостимулирующего и лютеинизирующего гормонов в крови приводит к фиброзу, гиалинозу и атрофии семенных канальцев. Яички перестают развиваться, становятся маленькими и плотными. Облитерация семенных канальцев заканчивается развитием азооспермии и бесплодия.
Распространённость заболевания
Синдром Клайнфельтера – наследственная патология, которая встречается примерно у 0,2% мужского населения. Проблема не диагностируется у девочек, так как сопряжена с наличием лишней хромосомы X у мальчиков. Данная мутация относится к числу часто встречающихся, так как регистрируется у одного младенца из 500. По распространенности синдром Кляйнтефера занимает третье место среди наследственных болезней у мужчин после сахарного диабета и гипертиреоза. При этом многие случаи патологии остаются не диагностированными. И даже с учетом этого факта данная проблема считается одной из самых часто встречающихся наследственных расстройств. Для сравнения, хорошо описанный в генетике синдром Амбраса встречается очень редко. На сегодняшний день известно лишь 50 случаев подтверждения врожденного гипертрихоза. А синдром Дауна, который также относится к распространенным генетическим патологиям, диагностируется лишь у одного младенца из 700.

Причины возникновения
Человеческое тело и психика — сложный механизм, который, удивительным делом, формируется из 23 пар хромосом. Любое нарушение ведет к отклонению в геноме, а следовательно и в организме, который будет сформирован на неправильной базе.
Синдром Клайнфельтера — это аномалия в формировании половых хромосом у мальчиков, а у девочек подобная болезнь обозначена как трисомия X. Заключается отклонение в том, что мужской стандарт XY получает дополнительные X или Y, за счет сбоя в яйцеклетке или сперматозоиде соответственно.
Синдром Клайнфельтера достаточно распространен в человеческой популяции, его частота: по разным данным — 1 из 500—800.
Клиника
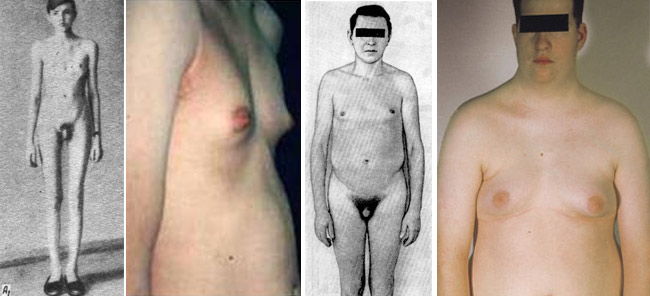
Мальчики с синдромом Клайнфельтера при рождении имеют нормальный рост и вес. Клинические признаки заболевания у них тоже отсутствуют: мошонка и половой член располагаются правильно и симметрично. Обнаружить патологию у новорожденных и грудных детей невозможно. Клиника синдрома Клайнфельтера становится явной только после начала пубертатного периода. Заподозрить заболевание можно и раньше по ряду характерных признаков. У больных мальчиков ноги длиннее, чем у сверстников, талия расположена несколько выше, мускулатура развита слабо. Узкие плечи и широкие бедра придают фигуре определенную женственность. Отложение жира также происходит по женскому типу. Диспропорциональное телосложение — один из характерных признаков данного наследственного заболевания.
Мальчики заметно прибавляют в росте между 6—7 годами. Большинство из них имеют атипичное строение лица. У больных детей часто заметно снижен интеллект и нарушено психическое развитие. Они не успевают в школе, быстро утомляются, плохо воспринимают устную речь, редко заводят друзей, избегают общения с незнакомыми людьми. Психика пациентов лабильна: часто возникают периоды полного равнодушия к происходящим событиям, чередуются радость и печаль, настроение меняется по малейшему поводу. Мальчики плохо адаптируется в социуме. Одни становятся скромными, замкнутыми, тихими и апатичными, другие — импульсивными и резкими, у третьих появляются склонности к криминалу.
У подростков развивается андрогенный дефицит, который проявляется определенными симптомами:
- У больных увеличиваются грудные железы. Развивается двусторонняя, безболезненная и необратимая гинекомастия, сохраняющаяся пожизненно. Ранняя гормонотерапия снижает выраженность гинекомастии. Половые гормоны необходимо принимать сразу после постановки диагноза.
- У большинства детей обнаруживают патогномоничный признак заболевания – маленькие и плотные яички. При этом половой член имеет размеры, не соответствующие возрасту. Сформированная мошонка часто без складок и пигментации. Простата не обнаруживается при пальпации. Гипогонадизм и гипогенитализм развиваются у всех больных.
- Волосы отсутствуют на лице и груди, растут на лобке треугольником. Вторичные половые признаки появляются очень поздно. Ухудшаются показатели спермограммы. Недоразвитие гортани проявляется высоким голосом у больных мужчин.
- Крипторхизм или неопущение яичек – врожденная аномалия, которая часто имеется у детей с синдромом Клайнфельтера. Мошонка становится асимметричной, в ней отсутствуют яички, возникает ноющая боль в паху.
- Неспецифические симптомы патологии — одышка, бледность кожи, гипергидроз, «жар» в теле, «приливы», покраснение шеи, кардиалгия, аритмия, гипертензия, сменяющаяся гипотензией, нарушение сна и аппетита, депрессивные признаки, отсутствие интереса к жизни, гипотонус кожи.
- Возникают боли в костях в результате остеопороза.

Инволюция тестикул сопровождается потерей фертильности. У молодых мужчин 20-25 лет присутствуют редкие поллюции, эрекция, сохраняется половое влечение. Ближе к 30 годам андрогенный дефицит становится максимально выраженным, что проявляется снижением либидо, уменьшением яркости оргазма и развитием импотенции. Взрослые пациенты часто становятся алкоголиками, наркоманами, гомосексуалами, особенно в условиях стресса. Ослабление полового влечения, нарушение половой функции и бесплодие – наиболее частые признаки, с которыми больные люди приходят к врачу.
Лица с синдромом Клайнфельтера часто страдают коллагенозами, гипо- или гипертиреозом, заболеваниями глаз, имеют аномалии скелета и пороки сердца. Если лечение патологии не начать вовремя, могут развиться тяжелые осложнения и печальные последствия. У больных нарушается психо-эмоциональное состояние, формируется умственная отсталость, появляются суицидальные наклонности, злоупотребление крепкими спиртными напитками, нарушается толерантность к глюкозе, кости становятся хрупкими, усугубляются врожденные пороки сердца, появляются новообразования молочных желез.
Многие случаи синдрома Клайнфельтера. остаются недиагностированными. Это приводит к отсутствию лечения, снижению качества жизни, инвалидизации больных, развитию остеопороза и сердечно-сосудистых заболеваний.
Как лечить синдром Клайнфельтера?
Полностью устранить представленную генную аномалию нельзя, поэтому терапия направлена на смягчение ее проявлений. Людям приходится постоянно купировать синдром Клайнфельтера – лечение требует пожизненного использования мужских половых гормонов, начиная с пубертатного периода (11-12 лет). Внутренний прием или инъекции тестостерона способствуют нормализации развития репродуктивной системы и ее функционирования.
Дополнительные способы лечения синдрома Клайнфельтера необходимы для облегчения сопутствующих проблем, которые провоцирует описываемое заболевание. Они включают:
- закаливание;
- профилактику инфекций;
- физкультуру;
- консультации психотерапевта и логопеда;
- стабилизацию веса;
- мастэктомию (при необходимости);
- соблюдение сбалансированного рациона.
Борьба с бесплодием

Главная проблема мужчин, страдающих синдромом Клайнфельтера — бесплодие. Вовремя начатая гормонотерапия поможет вести полноценную половую жизни и избежать эректильной дисфункции, но не способна восстановить фертильность.
До недавнего времени пациенты с XXY синдромом считались полностью бесплодными. Однако для людей с таким диагнозом выходом могут стать новые технологии экстракорпорального оплодотворения (ЭКО). У мужчин с синдромом Клайнфельтера наблюдаются разные показатели спермограмм — есть пациенты, у которых вообще нет сперматозоидов, но есть и те, у которых они присутствуют в малом количестве. Для вторых успешно используется вспомогательный метод ИКСИ (интрацитоплазматическая инъекция сперматозоида), при котором сперматозоид вводится в яйцеклетку с помощью иглы. По медицинским данным, это дает возможность почти четверти мужчин с синдромом Клайнфельтера стать биологическими отцами.
Даже пациентам с азооспермией, полным отсутствием сперматозоидов в эякуляте, врачи сегодня дают шанс стать родителями. Новые исследования говорят о том, что зародышевые клетки могут находиться непосредственно в яичках больного. И в этом случае для получения сперматозоида проводится биопсия органа.
Также врачи предполагают, что разрушение яичка и последующие проблемы с фертильностью и эрекцией возникают в подростковом возрасте, в период полового созревания. Если же синдром диагностирован раньше, есть шанс взять сперму или ткань яичка и сохранить их с помощью криоконсервации до начала патологических изменений.
Пройдите тестВаш персональный IQ здоровьяПройдите этот тест и узнайте, во сколько баллов – по десятибалльной шкале – можно оценить состояние вашего здоровья.
Двойные аберрации
Большая частота хромосомных аномалий в материале выкидышей объясняет высокую частоту комбинированных аномалий в одном и том же зародыше. Напротив, у новорожденных комбинированные аномалии крайне редки. Обычно в таких случаях наблюдаются комбинации аномалии половой хромосомы и аномалии аутосомы.
В связи с более высокой частотой аутосомных трисомий в материале выкидышей, при комбинированных хромосомных аномалиях у абортусов чаще всего встречаются двойные аутосомные трисомии. Трудно сказать, связаны ли такие трисомии с двойным «нон-дисджанкшн» в одной и той же гамете, или со встречей двух аномальных гамет.
Частота сочетаний различных трисомий в одной и той же зиготе носит случайный характер, что позволяет предположить независимость друг от друга появления двойных трисомий.
Комбинация двух механизмов, приводящих к появлению двойных аномалий, позволяет объяснить появление других аномалий кариотипа, встречающихся при выкидышах. «Нон-дисджанкшн» при образовании одной из гамет в сочетании с механизмами образования полиплоидии объясняет появление зигот с 68 или 70 хромосомами. Сбой первого митотического деления у такой зиготы с трисомией может приводить к таким кариотипам, как 94,XXXX,16+,16+.
Синдром Клайнфельтера – причины возникновения
Пока не установлено, почему у некоторых мальчиков появляется лишняя хромосома в половой паре. Есть только теории, из-за чего может возникать синдром Клайнфельтера – причины, предположительно провоцирующие аномалию:
- вирусные инфекции;
- слишком ранняя или поздняя беременность;
- кровное родство между отцом и матерью;
- неблагоприятная экология.
Синдром Клайнфельтера не передается по наследству. Присутствие одной или нескольких хромосом в половой паре в большинстве ситуаций вызывает бесплодие в зрелости. Больной рассматриваемой патологией не способен иметь наследников с аналогичной генетической мутацией. Лишняя хромосома может иметь как отцовское, так и материнское происхождение, но у женщин она обнаруживается чаще (67% случаев).
Диагноз
Диагноз до периода полового созревания может быть поставлен только при определении полового хроматина или кариотипа. После пубертатного периода возможен предположительный клин, диагноз на основании появления характерных симптомов, выявления повышенного выделения гонадотропинов, а при биопсии яичек — атрофии и гиалиноза семенных канальцев, узловой гиперплазии интерстициальных клеток.
Определение полового хроматина (см.) и изучение кариотипа (см.) являются основными методами дифференциального диагноза К. с. с другими синдромами гипогонадизма (см.), в частности с герминативной аплазией (см.).
Тактика общения с пациентами и их родителями
Важным аспектом ведения пациентов с синдромом Клайнфельтера является общение как с самими пациентами, так и с их родителями. Можно определить основные два направления в решении вопросов, возникающих при таком общении
Во-первых, как правильно информировать родителей пациентов о наличии у ребёнка синдрома Клайнфельтера и чему стоит уделить особое внимание. И во-вторых, необходимость полного информирования самих пациентов об этом синдроме и возможных его последствиях.
Необходимым условием для общения с пациентами и их родителями является, в первую очередь, правильная осведомленность врача о данном синдроме, другими словами, компетентность врача в данном вопросе.
Во многих странах синдром Клайнфельтера часто диагностируется ещё до рождения ребёнка, так как многие женщины позднего детородного возраста, в связи с высоким риском генетических дефектов у будущего потомства, используют пренатальную генетическую диагностику плода. Нередко пренатальное выявление синдрома Клайнфельтера является поводом для прерывания беременности, в том числе и по рекомендации врачей.
В России крайне редко проводится анализ кариотипа будущего ребёнка. Поэтому диагноз, как правило, устанавливается уже после рождения, а точнее в постпубертатном возрасте, когда начинают проявляться симптомы гипогонадизма, и именно в этот период отмечается наиболее частая обращаемость пациентов и их родителей к специалистам.
Сравнительная клиническая характеристика различных видов анеуплоидий
45,X/46,XY
При мозаицизме (46,XY/47,XXY) клинические симптомы выражены слабо, и отдельные больные могут сохранять, хотя и сниженную, способность к оплодотворению. Таким образом, при исследовании эякулята у пациентов с мозаицизмом могут обнаруживаться нормальные сперматозоиды, в отличие от немозаичных форм при генотипе 47XXY, или при более высокой степени анеуплоидий половых хромосом.
48, XXYY
Мужчины с кариотипом 48, XXYY отличаются более высоким ростом, обычно превышающим 182 см. Остальные клинические проявления ничем не отличаются от пациентов с кариотипом 47,XXY.
Что касается психологических особенностей, то обычно такие пациенты характеризуются как тихие и скромные, однако могут быть агрессивными и импульсивными.
В исследовании, сравнивающем 16 мужчин с кариотипом 48, ХХYY с 9 мужчинами, имеющими кариотип 47,XXY в возрасте 5—20 лет, было отмечено, что первая группа мужчин имеет более низкий показатель IQ, особенно за счёт снижения вербального компонента (коэффициент IQ находится в диапазоне 60-80). Речь у таких больных обычно замедлена. 48, XXYY мужчины являются также более склонными к агрессивному поведению и депрессиям по сравнению с мужчинами с кариотипом 47, XXY. К тому же у них отмечаются гораздо более низкие адаптивные возможности в социальной среде.
48, XXXY
Мужчины с кариотипом 48, XXXY могут иметь как высокий, так и средний рост. Часто отмечаются такие аномалии, как глазной гипертелоризм, плоская переносица, лучелоктевой синостоз, клинодактилия пятого пальца. Коэффициент интеллекта обычно находится в пределах 40—60, речь таких больных значительно замедлена. В поведении отмечается выраженный инфантилизм, который совместим с уровнем IQ. Таких мужчин обычно описывают как пассивных и не особенно агрессивных.
49, XXXXY
Пациенты с кариотипом 49, XXXXY имеют более выраженные нарушения физического и умственного развития. Они проявляются микроцефалией, глазным гипертелоризмом, плоской переносицей, узкими глазными щелями.
Рост таких больных обычно низкий. Они могут также иметь
расщеплённый нёбный язычок, волчью пасть, пороки сердца (в том числе открытый ductus arteriosus), лучелоктевой синостоз, вальгусное искривление коленных суставов, деформацию стоп, клинодактилию пятого пальца. Объём яичек, а также размер полового члена у таких пациентов маленькие. IQ снижен и находится в пределах 20 — 60. Их обычно описывают как скромных и дружелюбных, со случайными приступами раздражительности и вспышками гнева, имеют трудности в адаптации к изменяющимся условиям социальной среды.
Тактика общения с пациентами и их родителями
Важным аспектом ведения пациентов с синдромом Клайнфельтера является общение как с самими пациентами, так и с их родителями. Можно определить основные два направления в решении вопросов, возникающих при таком общении
Во-первых, как правильно информировать родителей пациентов о наличии у ребёнка синдрома Клайнфельтера и чему стоит уделить особое внимание. И во-вторых, необходимость полного информирования самих пациентов об этом синдроме и возможных его последствиях.
Необходимым условием для общения с пациентами и их родителями является, в первую очередь, правильная осведомленность врача о данном синдроме, другими словами, компетентность врача в данном вопросе.
Во многих странах синдром Клайнфельтера часто диагностируется ещё до рождения ребёнка, так как многие женщины позднего детородного возраста, в связи с высоким риском генетических дефектов у будущего потомства, используют пренатальную генетическую диагностику плода. Нередко пренатальное выявление синдрома Клайнфельтера является поводом для прерывания беременности, в том числе и по рекомендации врачей.
В России крайне редко проводится анализ кариотипа будущего ребёнка. Поэтому диагноз, как правило, устанавливается уже после рождения, а точнее в постпубертатном возрасте, когда начинают проявляться симптомы гипогонадизма, и именно в этот период отмечается наиболее частая обращаемость пациентов и их родителей к специалистам.
Последствия лечения синдрома Клайнфельтера
В целом прогноз жизни больных с синдромом Клайнфельтера благоприятный. Они имеют нормальную продолжительность жизни и при проведении заместительной терапии могут спокойно адаптироваться в обществе. Серьезными негативными моментами являются предрасположенность к развитию хронических заболеваний и бесплодие.
Возможность получения потомства
Ранее считалось, что больные с синдромом Клайнфельтера неспособны иметь потомство. Но на сегодняшний момент эта точка зрения пересмотрена. Причиной является развитие экстракорпорального оплодотворения и получение данных о том, что в яичках больных присутствуют зародышевые клетки. Предпринимаются попытки получения генетического материала непосредственно из яичка больного, при этом такой метод используется даже у мужчин с азооспермией. Есть удачные попытки оплодотворения яйцеклеток этими сперматозоидами.
Что такое синдром Клайнфельтера?
Синдром Клайнфельтера (СК) — заболевание, возникающее у мужчин, когда у них появляется дополнительная Х-хромосома. Некоторые мужчины с расстройством не имеют явных признаков или симптомов, в то время как другие могут иметь различную степень когнитивных, социальных, поведенческих и познавательных нарушений. Взрослые с синдромом Клайнфельтера могут также иметь первичный гипогонадизм (снижение выработки тестостерона), маленькие и/или неопущенные яички (крипторхизм), увеличенную грудь (гинекомастия), высокий рост и/или неспособность иметь детей (бесплодие), а также аномальное отверстие полового члена (гипоспадия) и маленький пенис (микропенис). Синдром Клайнфельтера не является наследственным расстройством, но обычно происходит как случайное событие во время формирования репродуктивных клеток (яйцеклеток и сперматозоидов), что приводит к наличию в каждой клетке одной дополнительной копии Х-хромосомы (47, XXY). Лечение состояния основано на признаках и симптомах, присутствующих у каждого пациента. Ожидаемая продолжительность жизни, как правило, нормальная, и многие больные с заболеванием имеют нормальную жизнь. Существует очень маленький риск развития рака молочной железы и других заболеваний, таких как хроническое воспалительное заболевание, называемое системной красной волчанкой.
В некоторых случаях в каждой клетке имеется более одной Х-хромосомы (например, 48, XXXY или 49, XXXXY). Эти состояния, которые часто называют «вариантами синдрома Клайнфельтера», обычно ассоциируется с более серьезными проблемами (умственная отсталость, проблемы со скелетом и нарушение координации), чем классический синдром Клайнфельтера (47, XXY).
Лечение
Синдром Клайнфельтера — распространенная и неизлечимая болезнь, которая имеет сглаженную картину проявления.
У многих мужчин данный симптом диагностируется лишь в зрелом возрасте, когда возникают ощутимые проблемы с либидо или продолжением рода. Как и в случае с синдромом Каллмана, поправить картину возможно с помощью пожизненной терапии гормонами. И чем раньше она начинается, тем меньше влияние синдрома на жизнь и здоровье мужчины.
Бесплодие и гинекомастия — необратимы даже с помощью гормонов. Однако, разработан протокол по забору спермы из яичек, который может привести к успешному искусственному оплодотворению. Болезнь Клайнфельтера — не наследственная и ее носитель «родит» здоровое потомство.
Что касается гинекомастии, то она удаляется только методами пластической хирургии. Вот пример успешной операции:
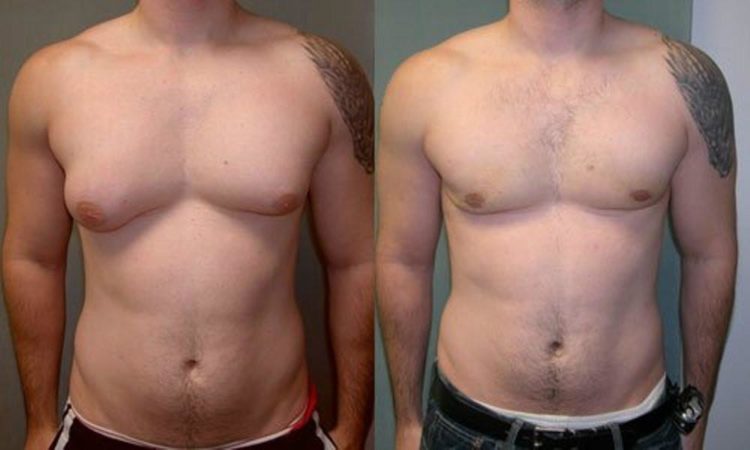
Большинство носителей аномального набора половых хромосом — нормальные люди, которые имеют шанс и право на полноценную жизнь. Лишняя генная информация не должна мешать им реализовать себя, завести семью. Информация про синдром обязана быть в широком доступе — до начала проблем, а не после.
Другим распространенным врожденным заболеванием у мужчин является гипогонадизм.
Про внешние проявления гипогонадизма вы узнаете отсюда. А за более подробной информацией добро пожаловать сюда:
- что такое первичный и вторичный гипогонадизм?
- симптомы заболевания у мальчиков;
- официальные и народные методы лечения.
Близкие расстройства
Синдром Кальмана (Каллмана) — редкое наследственное заболевание, которое в основном, но не исключительно, поражает мужчин. Основными характеристиками синдрома Каллмана как у мужчин, так и у женщин являются отсутствие полового созревания и полная или частичная потеря обоняния. Отсутствие полового созревания отражает гормональный дисбаланс, вызванный отказом части мозга, известной как гипоталамус. У пациентов с синдромом Кальмана наблюдаются признаки маленьких гениталий, стерильных гонад, которые не могут продуцировать половые клетки (гипогонадизм), и потери обоняния (аносмия). Нарушение выработки гормонов, а также сперматозоидов и яйцеклеток вызывает задержку полового созревания, рост и бесплодие.