Мышечная атрофия
Содержание:
- 3.Симптоматика, диагностика
- Диагностика
- Генетические анализы на мутации муковисцидоза (CFTR) и мутации AZF
- «Взрослые не у дел»
- Классификация
- Симптомы разных форм болезни
- Но чуда ждать не стоит
- Печень сильно пострадала
- Причины
- Проявления
- Денис Хайруллин, папа Варвары
- 4.Диагностика и лечение синдрома мальабсорбции
- «У меня огромный том переписки с нашими ведомствами»
- Суды и сборы
- Сколько стоят препараты?
- Промежуточные данные 3-й фазы STR1VEпо состоянию на 27 сентября 2018 г. (дата среза данных для анализа)
3.Симптоматика, диагностика
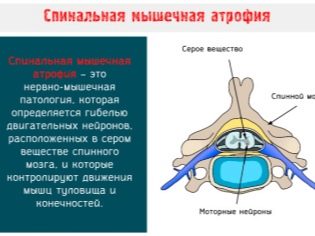
Заболевание начинается, как правило, с мышечной слабости и парезов (частичных параличей) верхних конечностей, постепенно распространяясь на таз и ноги. Зачастую отмечаются непроизвольные подергивания и сокращения мышц языка, лица и плечевого пояса, пальцевый тремор в позе Ромберга. Характерным для любой спинальной мышечной атрофии является также снижение тактильной чувствительности. Бульбарный компонент симптомокомплекса заключается в нарушениях и постепенном угасании глотательного, небного, жевательного рефлексов, затрудненной речи, нарастающей дыхательной недостаточности. Нередко процесс поражает также гипоталамус, что обусловливает эндокринные расстройства: так, у мужчин может начаться гинекомастия (увеличение грудных желез по женскому типу), бесплодие, атрофия яичек.
Учитывая достаточную специфику клинической картины, опытный невролог может установить диагноз СБМА клинически, – на основании осмотра, жалоб и анамнеза. Для уточняющего обследования назначается МРТ, электромиография (метод диагностики функционального статуса мышечной системы).
Диагностика
Диагноз СМА подозревается при наличии симптомов, а подтверждается молекулярно-генетическим тестированием. Молекулярно-генетическое тестирование используется для определения наличия мутации в гене SMN1. Типы СМА 0, 1, 2, 3 и 4 вызваны частичной или полной потерей гена SMN1, и около 95% затронутых пациентов будут демонстрировать делецию обеих копий определенной части (экзона 7 или экзона 8) гена. У около 5% пострадавших обнаружится делеция экзона 7 в одной копии гена SMN1 и другая мутация в другой копии гена SMN1. Молекулярно-генетическое тестирование также можно использовать для определения количества копий гена SMN2.
До появления молекулярного тестирования для диагностики использовались нейрофизиологические исследования и биопсия мышц, но эти тесты сегодня редко используются, если только нет проблем с молекулярно-генетическим тестированием.
Генетические анализы на мутации муковисцидоза (CFTR) и мутации AZF
При тяжёлых формах мужского бесплодия (азооспермии или значительной олигозооспермии, менее 5 млн сперматозоидов в 1 мл спермы неоднократно) врачи-андрологи обычно назначают дополнительные генетические анализы на мутации муковисцидоза (CFTR) и мутации AZF.
При выявлении носительства муковисцидоза рекомендовано обследовать и партнершу на это же заболевание (помним, что при наличии у партнеров одинаковой мутации, вероятность рождения больного ребенка у пары — 25%).
При выявлении мутации AZF рекомендовано ЭКО с ПГТ-А и перенос эмбрионов женского пола, так как мальчик унаследует такую же мутацию (Y хромосома неизбежно достанется ему от больного отца), и ребенок тоже будет бесплодным.
«Взрослые не у дел»
Летом 2020 года президент России Владимир Путин дал поручение создать фонд поддержки детей с тяжёлыми жизнеугрожающими и хроническими, в том числе редкими (орфанными), заболеваниями. Он будет финансироваться за счёт повышения НДФЛ до 15% для тех, чьи доходы составляют более 5 млн рублей в год, а также из частных пожертвований.
Согласно проекту постановления правительства РФ, в 2021 году в фонд будет выделена субсидия в размере 60 млрд рублей. Это позволит оказать помощь 25 тыс. пациентов.
Критерии отбора заболеваний и категорий детей будет разрабатывать экспертный совет, а попечительский совет займётся утверждением бюджета, перечня закупаемых лекарств и медицинских изделий, а также станет следить за деятельностью фонда.
По информации «Фармацевтического вестника», председателем правления должен стать протоиерей Санкт-Петербургской епархии, председатель Комиссии Общественной палаты по вопросам благотворительности и социальной работе и основатель некоммерческого учреждения «Детский хоспис» Александр Ткаченко.
Ольга Германенко называет создание фонда «беспрецедентным решением», однако отмечает, что ряд проблем ещё не преодолён: «Прекрасное решение с фондом и детской когортой в то же время дробит сообщество на группы. Взрослые пациенты — а их в нашем реестре более 200 человек — остаются как бы не у дел».
По её словам, в 2020 году в России стали расти интерес и осведомлённость в теме СМА, что, возможно, побудит пары делать генетические тесты до или во время беременности.
«Наши высказывания начинают обсуждать на разных площадках, в том числе на тех, где принимаются решения, — говорит Ольга Германенко. — И мы надеемся, что следующий год принесёт нам хотя бы пилотные региональные программы по тестам на СМА во время беременности или неонатальному скринингу. Это позволит рано выявлять болезнь и начинать лечение ещё до появления первых симптомов. Тогда дети — носители «сломанного» гена вырастут ничем не отличающимися от остальных детей».
Классификация
Самой распространенной формой СМА у детей является проксимальная. Она представлена несколькими видами заболевания, не все из которых становятся очевидными сразу после рождения ребенка.
- Болезнь Вердинга-Гоффмана – СМА 1 типа, тяжелая младенческая болезнь, которая проявляется в первые полгода жизни ребенка. Прогнозы при ней самые неблагоприятные, большинство пациентов погибают. Ребенок с СМА 1 типа не может ни стоять, ни сидеть, ни переворачиваться самостоятельно. У многих новорожденных нарушены сосательные и глотательные рефлексы. Часто отсутствует возможность самостоятельного дыхания или дыхание затруднено.
- Атрофия Дубовица – СМА 2 типа, поздняя младенческая. Проявляется обычно в возрасте от полугода до полутора лет и позднее. Ходить, стоять ребенок не может, но способен сидеть, питание не нарушено, он вполне справляется с задачей глотания, сосания. Сколько проживет малыш, зависит от того, в каком состоянии находятся дыхательные мышцы.
- Атрофия Кугельберга-Веландер – СМА 3 типа, инфантильная. Обычно обнаруживается в возрасте от полутора лет, чаще в два года. Прогностически более благоприятная форма. Маленькие пациенты могут стоять, сидеть, перемещаться, но испытывают сильнейшую слабость, а потому в большинстве случаев нуждаются в инвалидном кресле, без которого нормальная жизнедеятельность для них затруднена.
- Атрофия Кеннеди – СМА 4 типа, бульбоспинальная. Обычно считается взрослой формой, но изредка выявляется и у детей после 15 лет. На длительность жизни влияет редко, ослабление мышц происходит медленно, постепенно, человек, который вел обычную жизнь и считал себя здоровым, со временем становится инвалидом и утрачивает способность перемещаться самостоятельно.
У детей регистрируются не только изолированные формы СМА, когда, кроме дистрофии мышц, ничего не беспокоит, но и сочетанные формы, когда спинальная атрофия – не единственный диагноз и у ребенка есть другие генетические или врожденные проблемы, например, пороки сердца и сосудов, олигофрения.
Симптомы разных форм болезни
Существует общий набор признаков СМА, по которым можно заподозрить патологию, если других проблем не обнаруживается, либо диагноз вызывает сомнение. Группу симптомов сводят к проявлению вялого периферического паралича:
- выраженная мышечная слабость или атрофия разных мышечных групп;
- сначала в процесс вовлекаются конечности – симметрично, ноги, а затем руки, постепенно втягивается и туловища;
- отсутствуют расстройства чувствительности и тазовые нарушения;
- наиболее выраженные проблемы затрагивают проксимальные или дистальные мышечные группы.
У пациентов появляются подергивания и фибрилляции – мерцательные аритмии.
Признаки СМА1
Заболевание Верднига-Гоффмана бывает 3 видов:
- Врожденная форма. Начинается в течение 1-6 месяца жизни, обладает тяжелыми симптомами. Обнаружить признаки можно во внутриутробном развитии – эмбрион будет мало двигаться. Гипотония наблюдается сразу же после рождения ребенка. Такие младенцы не держат голову, не могут сидеть. Постоянно находятся в позе лягушки с раздвинутыми конечностями. Сначала симптомы появляются в ногах, затем в руках, после этого страдает дыхательная мускулатура. Психическое развитие у таких детей медленное, они редко доживают до 2 лет.
- Ранняя спинальная мышечная атрофия. Первые признаки начинают беспокоить пациента до 1,5 лет, чаще всего – после какой-либо инфекции. Даже если раньше ребенок мог стоять и сидеть, теперь он утрачивает эти функции. Развиваются парезы, а затем поражаются дыхательные мышцы. Ребенок умирает, как правило, в результате затяжной пневмонии или дыхательной недостаточности в возрасте 3-5 лет.
- Поздняя форма. Патология встречается после 1,5 лет, двигательные возможности сохраняются у ребенка до 10 лет. Медленный прогресс симптомов приводит к дыхательной недостаточности и смерти в возрасте до 18 лет.
Признаки болезни Кугельберга-Веландера
Возникает в возрасте от 2 до 15 лет. Сначала в процесс вовлекаются нижние конечности, затем пояс таза, на последних стадиях страдает плечевой пояс и дыхательная система. Примерно у 25% пациентов появляется синдром псевдогипертрофии мышц, из-за чего патологию путают с мышечной болезнью Беккера.
Спинальная мышечная атрофия Кугельберга-Веландера не сопровождается костными деформациями, и пациенты способны сами себя обслуживать в течение долгих лет.
Амиотрофия Кеннеди
Эта патология входит во взрослую группу, болеют представители мужского пола после 30 лет. Женщины от патологии не страдают. Течение – умеренное, сначала поражаются ножные мышцы, последующие 10-20 лет пациент сохраняет привычный ритм жизни. Только после этого начинают страдать мускулы рук и головы. У многих пациентов со временем наступают эндокринные изменения: атрофия яичек, отсутствие либидо, сахарный диабет.
Дистальная СМА
Эта форма спинально-мышечной атрофии также развивается у взрослых пациентов после 20 лет. Ее второе название – СМА Дюшенна-Арана. Риск развития патологии сохраняется вплоть до 50 лет. Атрофия начинается в руках, вызывает синдром «когтистой лапы», потом переходит на крупные мышцы. Со временем появляются парезы мышц нижних конечностей, а туловище страдает редко. Прогноз у этой формы благоприятный, если не присоединяется торсионная дистония или болезнь Паркинсона.
СМА Вюльпиана
Скапуло-перонеальная форма спинально-мышечной атрофии, сопровождаемая симптомом «крылатых» лопаток. Появляется в среднем в 20-40 лет, позже встречается реже. Поражается плечевой пояс, а через некоторое время – руки и нижние конечности. При этой форме спинальной болезни двигательные функции у пациента сохраняются на 30-40 лет.
Но чуда ждать не стоит
Многие родители детей со СМА считают «Золгенсму» чудом, которое может полностью вылечить их детей. При такой фантастической стоимости и уникальности лекарства их, конечно, можно понять. Препарат начал применяться в середине 2019 года сначала в США, потом в Европе. В России «Золгенсма» не зарегистрирована, но по жизненным показаниям разрешается к применению.
Принцип работы «Золгенсмы» такой: вирус-вектор, как грузовик, доставляет в клетку белок SMN1. Именно из-за того, что этот белок не вырабатывается, гибнут мотонейроны и не работают мышцы. Вирус, который использует для доставки «Золгенсма», непатогенный, он просто позволяет проникнуть внутрь клетки, но организм может вырабатывать антитела в ответ на его попадание — именно поэтому «Золгенсма» вводится один раз и не может вводиться повторно.
Препарат «Золгенсма» пока одобрен только для применения у детей до двух лет. При этом результаты клинических исследований есть только по группе для детей до шести месяцев. Обширных данных об эффективности для детей старшего возраста пока нет. Это дало право крупнейшему специалисту по СМА в Европе — профессору Лорану Сервэ говорить, что препарат эффективен только для детей младше 7 месяцев.
Фото: hellohope.com
— Если говорить в общих чертах, после инъекции «Золгенсмы» дети с досимптомным течением болезни развиваются нормально, — объясняет Лоран Сервэ. — Трехмесячные пациенты с постсимптомным течением болезни чувствуют себя значительно лучше, но все равно далеки от нормы. А для пациентов старше полугода данных об эффективности препарата нет.
По словам ученого, сейчас проводится два исследования: досимптомное и постсимптомное. Но даже в постсимптомный период препарат вводят только детям в возрасте до шести месяцев, потому что после этого возраста вероятность, что лекарство подействует, невелика. Пациенты, получившие лекарство тогда, когда симптомы заболевания уже проявились, чувствуют себя лучше, но далеки от выздоровления.
— Существует на данный момент три препарата для лечения СМА: «Спинраза», «Золегнсма» и «Рисдиплам», — объясняет Александр Поляков, профессор РАН, ведущий специалист в области молекулярной генетики и ДНК-диагностики наследственных заболеваний
— Неважно, какой препарат используется. Все эти препараты по-разному воздействуют на те мотонейроны, которые у больного остались живыми
Они дают возможность им работать. Соответственно, если мотонейроны умерли, то эффективности не будет. Не существует убедительных работ, которые бы говорили о большей эффективности того или иного препарата. Мы пока не знаем, хватит ли «Золгенсмы» до конца жизни. Самые длинные исследования сейчас — это 5-7 лет. Но так как «Спинразу» нужно вводить пожизненно, чтобы остановить прогрессирование болезни, в масштабах жизни «Золгенсма» пока получается дешевле.
В долгосрочном клиническом исследовании «Золгенсмы», ставшем одним из основных при одобрении препарата, участвовали 15 пациентов. Трое из них получали низкую дозу, а 12 — терапевтическую. Из этих двенадцати два пациента стоят самостоятельно и ходят, десять — сидят. Они никогда не сели бы сами, без лечения. Один пациент не смог сидеть самостоятельно. Но он чувствует себя хорошо, не нуждается в вентиляции легких, удерживает голову. Те больные, которые начали ходить, оба получили терапию в возрасте до трех месяцев.
Что такое Золгенсма. Главное — о самом дорогом в мире лекарстве
— Да, вокруг этого препарата сейчас много сборов и много дискуссий, — говорит Ольга Германенко, директор благотворительного фонда «Семьи СМА». — У людей, которые не очень разбираются в нюансах, складывается впечатление, что этот препарат является чудесным средством, которое действительно способно полностью вылечить ребенка со СМА. Это, к сожалению, не так. Это генная терапия, она влияет на первопричину заболевания, и многие думают, что она берет тот участок ДНК, который пострадал и является причиной болезни, и что-то в него докладывает, то есть чинит ДНК. Это тоже не так. Этот препарат доставляет отсутствующий белок в клетку человека. Но лекарство ничего не делает с ДНК, и этот белок не восстановит клетки, которые уже умерли. То есть действовать он, по сути, будет так же, как «Спинраза»,— чем раньше применили, тем лучше, а если человек потерял двигательные нейроны, то лекарство их уже не вернет.
Печень сильно пострадала
Наиболее частые нежелательные реакции на фоне применения «Золгенсмы» — это повышение активности печеночных ферментов. В инструкции по применению препарата есть предупреждение о риске развития тяжелого острого поражения печени. Перед назначением препарата необходимы клинический осмотр и лабораторная оценка функции печени, а после введения нужен мониторинг печеночных нарушений в течение по меньшей мере 3 месяцев.
— У Лизы печень очень сильно пострадала после «Золгенсмы», — говорит папа Лизы Краюхиной, — шесть месяцев она восстанавливалась, но дочь справилась.
Семимесячному Мише Бахтину «укол жизни» сделали совсем недавно — 19 февраля 2021 года.
— Очень сильно печень реагирует на препарат, — говорит папа Миши, Дмитрий. — На сегодняшний день у Миши печеночные показатели завышены, но врачи увидели стабильность в показаниях и выписали домой. И это, конечно, правильное решение. Идет еще гормональная терапия, иммунитет подавлен, и лучше быть дома, чтобы не хватать больничные инфекции.
Причины
Как уже говорилось, речь идет о генетическом заболевании, а потому причины его возникновения – область поисков генетиков. Ребенок наследует один из рецессивных генов на пятой хромосоме (это могут быть гены SMN, NAIP, H4F5, BTF2p44).
Вероятность передать потомству такой ген у носителя высока – 25%. Если и мама, и папа – скрытые носители мутировавшего гена, то вероятность СМА у ребенка – 50%. Пораженный аномальный ген не дает нормально протекать процессам выработки белка SMN и нервные клетки, отвечающие за двигательные функции мышц, в спинном мозге начинают постепенно погибать. Процесс их гибели продолжается и после того, как малютка появится на свет.
Проявления
Симптомы зависят от типа болезни. Поскольку мы рассматривает только детские четыре типа, то следует отметить, что для всех характерна мышечная слабость, мышечная атрофия. В остальном каждый тип имеет собственную клиническую картину и отличительные особенности.
- СМА 1 типа (атрофия Вердинга-Гоффмана) доступна для обнаружения еще во время беременности. Заподозрить заболевание у плода врач может при очень вялых шевелениях. Но подтвердить диагноз на этапе вынашивания ребенка сложно, это обычно происходит после родов. Малыш с такой атрофией не может сам держать головку, ворочаться с боку на бок, не садится. Он почти постоянно лежит на спинке, его поза расслаблена, он не поднимает ножки, не сводит их вместе, не складывает вместе ладошки рук. На самом раннем этапе могут быть огромные проблемы с тем, чтобы накормить ребенка, ведь глотать у него получается очень плохо или не получается. Большая часть деток погибает еще до двухлетнего возраста. Некоторым удается дожить до семи-восьми лет, но атрофия только усиливается. Обычно гибель происходит из-за недостаточности работы сердца, легких, пищеварительных органов.
- СМА 2 типа (атрофия Дубовица) при рождении обычно не обнаруживается, ведь ребенок способен дышать, глотать пищу, и только после полугода становится очевидным прогресс мышечной атрофии. Если первые симптомы приходятся на возраст, когда ребенок уже научился стоять в кроватке, то ярким признаком может быть подкашивание ножек, беспричинные падения крохи. Постепенно ему становится сложно глотать. Со временем ребенок начинает нуждаться в инвалидном кресле.
- СМА 3 типа (амиотрофия Кюгельберга-Веландера) может обнаружиться в любом возрасте после 2 лет до совершеннолетия. Ребенок, который нормально рос и развивался, постепенно начинает жаловаться на слабость, обычно в области плеч, предплечий. По мере прогрессирования ему становится трудно бегать, ходить по лестнице, приседать. Все зависит от ухода – некоторые сохраняют способность перемещаться самостоятельно на протяжении долгих лет.
- СМА 4 типа (атрофия Кеннеди) встречается только у пациентов мужского пола, поскольку считается сцепленной с половой хромосомой Х. Первые признаки – слабость в области бедренных мышц, постепенно поражаются черепные нервы. Заболевание прогрессирует медленно.
Денис Хайруллин, папа Варвары
— Мы познакомились с супругой 12 лет назад, оба мы из Благовещенска, сейчас живём в Уфе. Я работаю в сфере сотовой связи: начинал с позиции, теперь я руководитель розничной сети, у меня в подчинении 25 магазинов. Жена работала администратором, потом ушла в декрет. У нас есть старшая дочка Ева, ей 4,5 года. А 12 февраля 2019 года на свет появилась Варя.
Когда младшей дочке было девять месяцев, мы забили тревогу. В таком возрасте ребёнок должен уже вставать, а некоторые уже ходят. Варя не могла встать, ножки были очень слабые, так что мы начали ездить по больницам. Только летом 2020 года нас направили сдавать генетический анализ, и лишь в сентябре поставили диагноз. Мы потеряли много времени.
Когда мы начали узнавать что-то о СМА, то погрузились в кошмар, из которого, казалось, нельзя выбраться. Но мы сразу решили, что лечиться будем «Золгенсмой»: препарат вводится только один раз, к тому же два других лекарства, «Спинразу» и «Эврисди», у нас в регионе всё равно быстро не достать.
Сбор мы открыли 28 сентября 2020 года. Информацию о Варе помогали распространять родственники, друзья, потом присоединились незнакомые люди из Уфы и Благовещенска. По согласованию с администрациями этих городов мы провели автопробег, благотворительные ярмарки при поддержке спонсоров. Волонтёры в соцсетях пытались достучаться до российских звёзд, известных блогеров, писали в различные компании. Конечно, было много отказов, но неравнодушные люди всё равно не сдавались.
За два с половиной месяца нам удалось собрать чуть больше 10 млн рублей, этого мало для закупки лекарства. Параллельно мы собирали необходимые документы, чтобы подать заявку на участие в лотерее, которую устраивает производитель «Золгенсмы». Мы использовали все свои возможности. Где-то что-то должно было выстрелить.
С сентября у нас с женой не было ни одного выходного дня, мы физически очень устали, у нас почти опустились руки. И вот поздно вечером 10 января 2021 года супруге позвонила невролог Вари и, плача от радости, сообщила, что мы выиграли в лотерею.
Представьте, что у тебя на плечах был огромный камень, ты уже выбился из сил его тащить, уже готов упасть и не встать. А потом в один момент он у тебя слетает. Вот что мы почувствовали тогда.
Сразу после того, как мы узнали о выигрыше, объявили о закрытии сбора. Из собранных денег мы оставили 3 млн на дальнейшую реабилитацию и закупку вертикализатора. 6,9 млн мы перевели на счёт маленького «смайлика» Саши Горобинского. Около 442 тыс., собранных через второй наш фонд, отдали Матвею Куликовскому, «смайлику» из Санкт-Петербурга. Порядка 100 тыс. рублей, которые собрали на зарубежной площадке, перевели мальчику с трахеостомой из Уфы, а деньги, которые поступили на счёт после закрытия сбора, — это около 20 тыс. рублей — перевели девочке-«смайлику» из Барнаула.
7 февраля мы, отсидев карантин, чтобы Варя не заболела накануне госпитализации, прилетели в Москву. 11 февраля, уже после сдачи всех анализов, нашей дочке сделали инъекцию «Золгенсмы» в НИКИ педиатрии им. Вельтищева. На следующий день Варе исполнилось два года.
Дочка ещё слабенькая, изменения пока трудно заметить — слишком мало времени прошло. Через месяц показатели печени должны прийти в норму, и тогда будем смотреть на эффект.
Мы с женой понимаем, что впереди ещё много работы, ведь многое зависит от реабилитации. Помимо вертикализатора, купим инвалидное кресло, хотя сразу решили, что не будем к нему именно как к инвалидному относиться — это просто средство, чтобы Варя передвигалась куда захочет. Плюс она пройдёт месячную реабилитацию в центре нашего фонда «Мать и дитя».
Наша старшая дочь Ева раньше ходила в садик, но после того, как мы узнали диагноз Вари, пришлось забрать её оттуда. Детям со СМА противопоказано любое заболевание, а в садиках, сами знаете, малыши часто болеют. Поэтому сейчас Еву учит моя мама, она преподаватель русского языка и литературы.
Варя улыбчивая, позитивная и подвижная. Даже врачи говорят, что для такого диагноза наш ребёнок прямо живчик. И мы очень надеемся увидеть эффект от лекарства через месяц.
4.Диагностика и лечение синдрома мальабсорбции
Исследования при подозрении на СМА охватывают широкий спектр физиологических показателей. Необходимо не только подтвердить предполагаемый диагноз, но и, по возможности, выяснить причины и механизм возникновения имеющихся проявлений синдрома.
Врач тщательно изучает анамнез пациента (в том числе семейный) и жалобы, уточняет характер и тяжесть нарушений, факторы и условия, провоцирующие проявление патологии.
Лабораторная диагностика включает:
- анализ кала на паразитов, инфекции и скрытую кровь;
- общий анализ крови;
- биохимический анализ крови;
- копрограмму.
Дополнительные диагностические данные, позволяющие выработать индивидуальный лечебный план, могут быть получены путём инструментальных методов диагностики:
- УЗД;
- МРТ и КТ;
- Эзофагогастродуоденоскопия;
- Ректороманоскопия;
- Исследование желудочного биоматериала на Helicobacter pylori.
Лечение синдрома мальабсорбции всегда сугубо индивидуально и должно учитывать характер и тяжесть специфических и общесоматических проявлений. Назначение лечебных препаратов обычно сочетается со строгой диетой. Приводится терапия сопутствующих и фоновых заболеваний.Рекомендовано ограничение физических нагрузок и психоэмоциональный покой. В большинстве случаев необходима сбалансированная витаминотерапия. При наличии инфекции применяются антибиотики и антимикотики. В ряде случаев необходимы ферменты и гормональные препараты.
Больной в процессе лечения регулярно проходит промежуточную диагностику для получения сведений о динамике и адекватности проводимой терапии. Лечебный курс должен быть пройден комплексно. Даже при видимом улучшении нельзя прекращать приём препаратов, поскольку временное улучшение может смениться ещё более тяжёлым обострением.
«У меня огромный том переписки с нашими ведомствами»
Милана Семенькова
Милана Семенькова, 2,5 года, Калининград. Получила Золгенсму 15 июля 2020 года. Рассказывает мама Виктория Кашуба
– Милана – наш первенец. Мы были счастливы, когда она родилась, начали копить деньги на то, чтобы взять ипотеку, планировали свою жизнь.
Но в 8–9 месяцев мы заподозрили неладное, дочка была слабой, плохо развивалась. Милана была вяленькая, не вставала на ножки. А врачи говорили нам, что все хорошо. В год мы узнали о болезни: нам поставили диагноз – СМА 2 типа. Она хотя бы ползала, вставала на коленки. Дети с первым типом СМА вообще часто лежачие, и там все гораздо раньше становится понятно.
Сначала мы надеялись на Спинразу. Я написала во все службы, в Минздрав и так далее. Нам сказали – у нас денег нет, собирайте сами. Но уже появилась Золгенсма. Я поняла, что пожизненно собирать деньги на Спинразу нереально – первый курс стоит 45 млн рублей, а потом – 25 млн ежегодно. Наверное, надо решиться на сбор на Золгенсму.
Параллельно я вела 3 месяца переговоры с Минздравом, чтобы нам сделали укол Спинразы. И мы получили ее, когда уже закрыли сбор по Золгенсме, через 9 месяцев. У меня огромный том переписки с ведомством.
На препарат мы собрали в мае 2020 года. Вложили и все свои деньги, уже было не до ипотеки. Нам приходили пожертвования даже из других стран – Германии, Казахстана, Армении, Литвы, Польши. В Калининграде мы расставили ящики для пожертвований. Провели автопробег, благотворительный концерт.
Когда началась пандемия коронавируса, все мероприятия запретили, мы испугались, что не соберем нужную сумму, но, как ни странно, частных пожертвований стало даже больше. Помогали и звезды. Но крупных пожертвований, по нескольку миллионов у нас не было, собрали именно благодаря помощи обычных людей и небольших сумм.
К тому времени ушло все, что Милана хотя бы могла делать: сначала она пыталась вроде бы вставать, а к лету 2020 года уже еле сидела, только если обложить подушками.
Укол Золгенсмы сделали 15 июля 2020 года. За это время мы сделали лишь 3 укола Спинразы, но это были загрузочные дозы, я не видела никаких изменений. А пока мы ждали Золгенсму, силы у Миланы уходили.
А если бы мы не сделали этот укол, нам бы пришлось колоть Спинразу. Но это бесконечные сборы средств. Сейчас думаю, что если бы нам сразу ввели Спинразу, мы бы уже не открыли сбор средств на Золгенсму. Это очень тяжело эмоционально и очень подрывает силы, а ведь у некоторых родителей эти сборы идут и по полтора года.
После укола Золгенсмы мы видим серьезные изменения. Милана уже может стоять на коленях, сидит без поддержки, пытается ползти, сидя на попе, и таким образом передвигается. Кстати, СМА не влияет на интеллект, так что Милана рано стала говорить, и уже говорит предложениями, поет песни, читает стихи, очень умный ребенок.
Суды и сборы
По данным руководителя фонда «Семьи СМА» Ольги Германенко, по всему миру лечение «Спинразой» получили 10 тыс. человек. В России по состоянию на середину декабря начали или продолжили инъекции этого препарата 209 человек, из них восемь взрослых.
«Показатель хороший, но недостаточный», — констатирует она.
При этом, по словам Ольги Германенко, именно в 2020 году некоторые семьи пациентов со СМА столкнулись с затягиванием процесса назначения препарата, в том числе и через суд. Среди доводов региональных властей — неопределённость источников и порядка финансирования закупок, отсутствие «Спинразы» в перечне жизненно необходимых и важных лекарственных препаратов (ЖНВЛП), а также малая эффективность терапии.
24 ноября 2020 года «Спинраза» была внесена в перечень ЖНВЛП. Теперь государство может контролировать стоимость лекарств в перечне, а региональные минздравы лишились возможности отказывать в предоставлении дорогостоящего препарата под предлогом его отсутствия в списке.
Кроме того, производитель «Спинразы» выразил готовность снизить её цену на 25%, таким образом, каждый четвёртый пациент сможет лечиться за счёт этой экономии. Также на основании перечня ЖНВЛП формируются так называемые территориальные программы медицинской помощи — значит, регионам будет проще закладывать соответствующий бюджет.
Через два дня после включения «Спинразы» в перечень жизненно необходимых лекарств произошло ещё одно важное для всех пациентов со СМА событие: в России был зарегистрирован препарат «Эврисди». К тому же появление второго зарегистрированного препарата в перспективе может ещё снизить цены на рынке
К тому же появление второго зарегистрированного препарата в перспективе может ещё снизить цены на рынке.
Регистрация в России самого дорогого лекарства от СМА, «Золгенсмы», ожидается в 2021 году. Пока этого не произошло, семьи пациентов либо собирают деньги на лечение в частном порядке (единичные сборы даже удалось закрыть), либо судятся за право получить терапию за счёт государства.
Особенность «Золгенсмы» в том, что, согласно инструкции, препарат надо применить до того, как ребёнку исполнится два года, тогда как терапию «Спинразой» и «Эврисди» можно начать в любом возрасте.
Опираясь именно на этот довод, суд отказал в предоставлении «Золгенсмы» Косте Гепалову из Санкт-Петербурга, которому в сентябре исполнилось два года. Однако летом специалисты в ЕС, а с ними и американские медики пришли к выводу, что важнее критерий веса ребёнка, а не его возраста. Семья Гепаловых подала повторный иск в суд с новыми документами и ожидает решения.
Сколько стоят препараты?
Компании при определении цены новых препаратов ориентируются не на себестоимость (хотя в случае Золгенсмы она довольно высока и составляет $150 000 — $200 000), а на эффект, который терапия оказывает на пациентов и здравоохранение в целом.
Стоимость первого года лечения Спинразой установлена компанией Biogen в США в $750 000, последующих – в $325 000. В Европе цены примерно в полтора раза ниже, и государственные плательщики договариваются о дальнейшем снижении стоимости. На Золгенсму Novartis объявила цену в США $2,125 млн за одну инъекцию, что позволяет называть ее самым дорогостоящим лекарством в мире.
Novartis исходила из того, что уже через 5 лет стоимость лечения Спинразой превзойдет стоимость Золгенсмы. Однако надо заметить, что период наблюдения за пациентами пока слишком мал, чтобы говорить о долговременном превосходстве Золгенсмы. Компания готова сотрудничать с государственными и частными страховыми системами в разных странах над схемами оплаты препарата, чтобы сделать его доступным для пациентов.
Между тем, по подсчетам Института клинических и экономических обзоров США (ICER) Спинраза не должна стоить дороже $72 000 за годовой курс, а Золгенсма — $1,5 млн за инъекцию.
В декабре 2019 Novartis объявила, что раздаст 100 доз Золгенсмы бесплатно, используя принцип лотереи. Европейское пациентское сообщество SMA Europe подвергло компанию критике, упрекнув в неэтичности такого решения. Компания начала работу над улучшением выбранного подхода.
Материал подготовлен в рамках совместного проекта с сайтом о науке «Биомолекула«
Автор благодарит Ольгу Германенко, учредителя и директора фонда «Семьи СМА» за помощь в работе над текстом
Промежуточные данные 3-й фазы STR1VEпо состоянию на 27 сентября 2018 г. (дата среза данных для анализа)
STR1VE — это продолжающееся открытое простое многоцентровое исследование с однократной дозой, предназначенное для оценки эффективности и безопасности однократного внутривенного введения Zolgensmaу пациентов с СМА 1-го типа, которые на момент генной терапии не достигли возраста шести месяцев. Исследование было разработано для охвата как можно более широкой популяции пациентов с СМА 1-го типа с одной или двумя копиями резервного гена SMN2, которые имеют биаллельную делецию гена SMN1 или точечные мутации. Эти критерии хорошо соответствуют популяции пациентов, которые были зарегистрированы в основном испытании 1-й фазы START, при этом потенциально предоставляя лечение некоторым из более редких субпопуляций на исследовательской основе. Исследование STR1VEпланируется завершить в 2020 году.
По состоянию на 27 сентября 2018 года 21 из 22 (95 процентов) пациентов были бессобытийно живы (прим. «событие» смерть или необходимость в длительной вентиляции легких более 16 часов в сутки). Средний возраст составлял 9,5 месяцев, 6 из 7 (86 процентов) пациентов смогли достичь возраста в 10,5 месяцов или старше без событий на указанную дату. Естественная история развития заболевания без лечения показывает, что 50 процентов детей с СМА 1-го типа не выживут или будут нуждаться в постоянной вентиляции к возрасту 10,5 месяцев.
Оценки по шкале специального теста нервно-мышечных расстройств CHOP-INTEND увеличились в среднем на 7,0 баллов через месяц после применения терапии и на 11,8 баллов через три месяца, отражая улучшение двигательной функции по сравнению с исходным уровнем.
Эти данные сопоставимы с улучшениями CHOP-INTENDпри предложенной терапевтической дозе когорты (2-я когорта) в основном исследовании START, которое продемонстрировало среднее соответственное увеличение на 9,8 и 15,4 баллов на том же отрезке времени. Раннее увеличение баллов по CHOP-INTENDможет быть связано с возможным достижением моторных навыков пациентами.
Предварительные оценки пациентов, получавших Zolgensma, показали улучшение двигательных навыков. Три пациента могли сидеть без поддержки в течение не менее 30 секунд по состоянию на 27 сентября 2018 года (медиана возраста — 9,4 месяца) и восемь пациентов достигли того же рубежа к 31 декабря 2018 года (средний возраст — 12,5 месяцев).
| контрольная отметка, кол-во пациентов n (%) | 3 фаза исследования STR1VE, n=22 | |
| 27 сентября 2018 | 31 декабря 2018 | |
| Держит голову прямо 3 и более секунд без поддержки | 12 (54,5) | 17 (77,3) |
| Переворачивается со спины и в правую, и в левую стороны | 3 (13,6) | 7 (31,8) |
| Сидит без поддержки 30 и более секунд | 3 (13,6) | 8 (36,4) |
| Стоит с помощью | 1 (4,5) | |
| Средняя продолжительность наблюдения при сборе данных | 5,5 месяцев | 8,1 месяцев |
| Средний возраст при сборе даных | 9,4 месяцев | 12,5 месяцев |
| Пациенты старше 12 месяцев, кол-во n (%) | 5 (22,7) | 13 (59,1) |
Наблюдения за безопасностью сравнимы с наблюдениями в 1-й фазе ключевого исследования START. Неблагоприятные события, представляющие особый интерес, включая повышение уровня трансаминаз, снижение количества тромбоцитов и тромбоцитопения, были преходящими и не вызывали каких-либо долгосрочных последствий. Один пациент умер от дыхательной недостаточности, которая была признана исследователем и независимым советом по мониторингу безопасности данных не связанной с лечением. Этот пациент продемонстрировал значительное улучшение моторики, с увеличением CHOP-INTENDна 27 баллов по сравнению с исходным уровнем через пять месяцев после вливания.
AveXis благодарен мужественным пациентам и семьям, которые принимают участие в наших испытаниях, что позволяет нам продолжать прилагать усилия по осуществлению значительных изменений в жизнь пациентов с редкими генетические заболеваниями.